Compounded pharmaceutical dosage forms may be generally described as preparations that are not commercially available from major pharma corporations. These preparations have traditionally been prepared in 503A pharmacies for identified patients; larger quantities of compounded preparations are also prepared by 503B outsourcing facilities for commercial distribution (1). Numerous dosage forms are compounded, including investigational drugs, parenteral nutrition, oncology preparations, radiopharmaceuticals, veterinary medications, and replacement commercial products during drug shortages (2-3). Individual compounded dosage forms may be prepared by pharmacists in community or hospital settings; greater scale compounding may include formulation scientists, engineers, and QA personnel in multi-facility corporations. Compounding may be completely manual or automated and may utilize electronic systems (e.g., automated compounding (ExactaMix), programmable robots (Equashield Pro), or pharmacy management (DoseEdge)). Aside from patients and clients, compounders may have relationships with other compounding organizations; (e.g., 503A hospitals may purchase compounded preparations from 503B facilities, and 503B organizations may provide clinical supplies to pharma industry.
Discussion topics
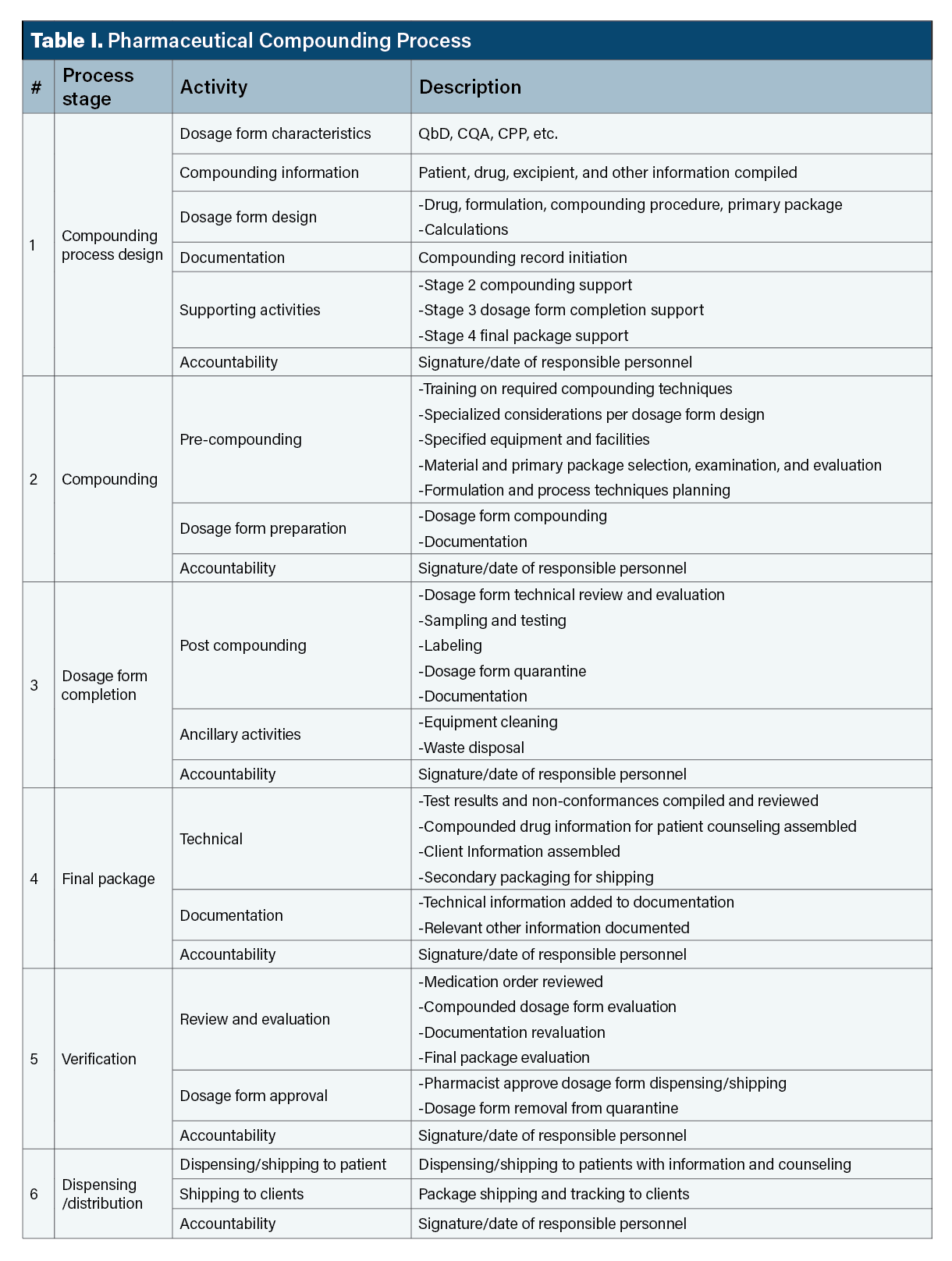
This discussion describes an ordered process for compounding pharmaceutical dosage forms in workplace environments. The process comprises six stages generally applicable to all compounding (Table I).The first four stages directly address dosage form preparation, evaluation, packaging, and medical information. Stage 5 evaluates and verifies previous completed stages; stage 6 comprises dispensing/shipping to patients or clients. Individual activities within stages will vary depending on the type of compounded dosage form (e.g., sterile vs. non-sterile). Activities in stages may be done by multiple people (e.g., pharmacists, technicians, engineers, etc.).
The compounding process was developed considering academic teaching of compounding theory and practice, problem experiences in multiple workplaces, published regulatory observations, and medication error incidents. Academic compounding typically focuses on placebo formulations and techniques in a classroom or laboratory. Compounding in the workplace is much different—active drugs with specific and sometimes incompatible physicochemical properties, actual patients, emergency circumstances, imperfect facilities, incomplete information, inadequate staffing, excessive workloads, and logistics with interruptions, interferences, and other distractions. Basic activities in respective compounding stages are described; potential problems are identified; technical science, compliance, documentation, and personal responsibility are emphasized. The process builds on pharmaceutical and regulatory concepts, including Quality by Design (QbD) (4-6), Quality Risk Management (QRM) (7), and Pharmaceutical Quality Systems (8,9). Concepts described by professional pharma organizations are included (10-13). The technical responsibilities of management supporting compounding are also addressed.
This discussion assumes organization compliance with USP, federal, state, local, other regulatory, and professional certifications; 503B outsourcing facilities must also comply with pharmaceutical Good Manufacturing Practices (GMP). It also assumes availability of approved policies and standard operating procedures (SOPs) at the site. Non-technical considerations, such as drugs and supplies inventory, third-party reimbursement, personnel development, performance metrics, and other business responsibilities, are not addressed.
Stage 1 compounding process design
Stage 1 comprises identification of all activities required to completely prepare the compounded dosage form and its associated final package. This includes formulation ingredients, preparation, testing, and other post-compounding activities for dispensing to patients or shipping to clients. Relevant information is integrated in stage 1. When stage 1 is completed, all activities for future performance will have been documented to guide the compounding process. Actions to minimize potential hazards (i.e., what might go wrong) are incorporated in the process. Stage 1 design provides technical and compliance consistency throughout all compounding activities. Stage 1 design activities are executed in stages 2, 3, and 4.
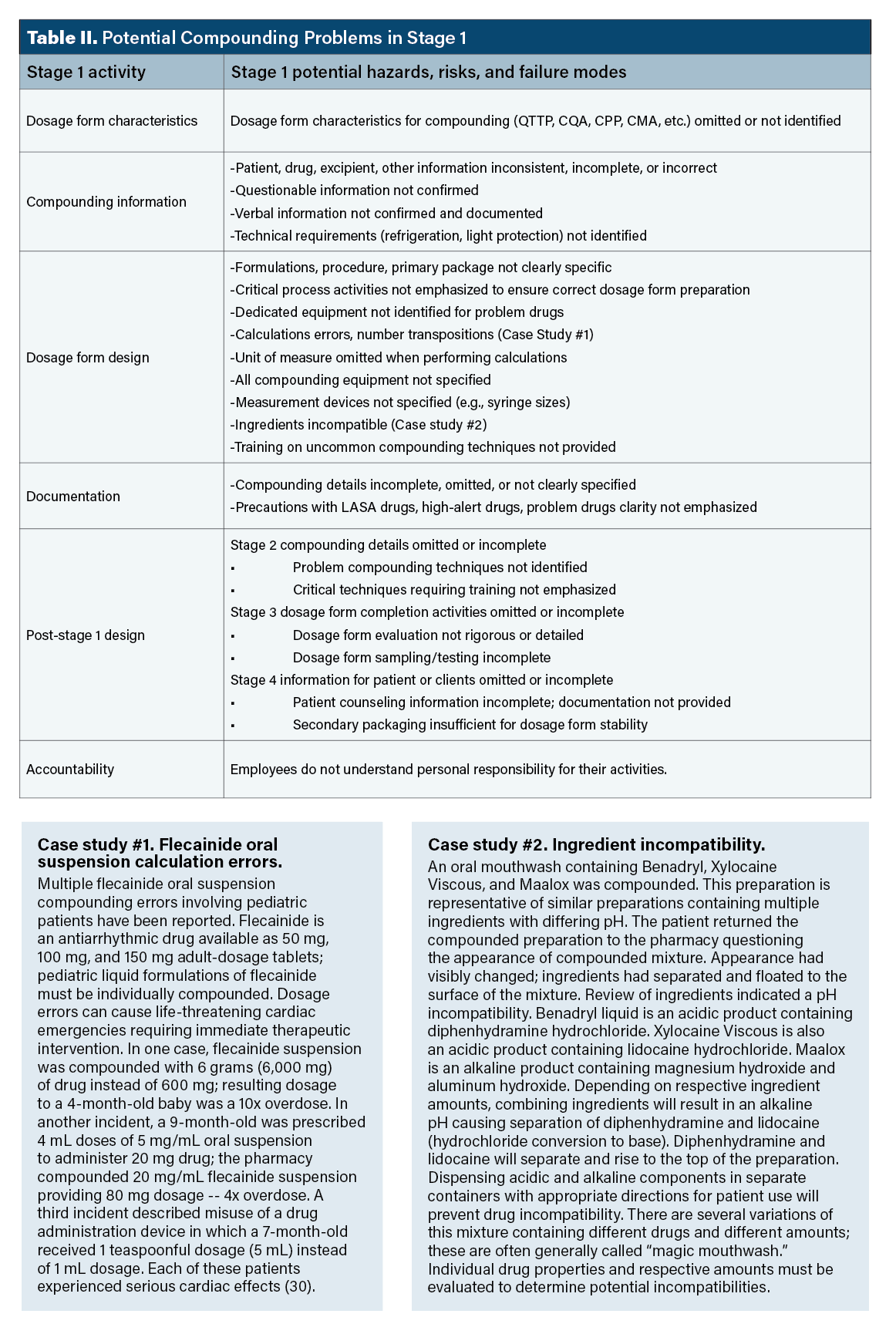
Dosage form identification and characterization. The first significant activity in the compounding process comprises evaluation of the medication order for the compounded dosage form. This includes determining the availability of commercial product fulfilling the medication order and making compounding unnecessary; using a commercial product is preferable to extemporaneous compounding. Compounded dosage forms may not be reimbursed by a third party if similar products are commercially available. If a suitable commercial product does not exist, the compounded dosage form is identified. Objectives analogous to QbD for commercial products, such as Critical Quality Attributes (CQA) and Critical Process Parameters (CPP), are noted. These are straightforward for an IV solution with a single additive; other compounding (e.g., parenteral nutrition, non-sterile formulations, etc.) are much more complex. Clear directions for the dosage form to be compounded are essential.
Compounding information. Concurrent with dosage form identification is verification of information relevant to compounding. Questionable, inconsistent, or otherwise problematic information must be corrected or confirmed. Technical information may be applicable throughout the compounding process (e.g., a light-sensitive drug must be correctly handled during preparation, post-compounding, packaging, shipping, and administration to the patient). Using an investigative approach can be helpful when presented with new compounding—what is known, what is not known and must be determined, and what is the desired outcome? In other words, where do you want to go and what do you need to know and do to get there? This approach will provide guidance and direction for the compounding effort. Missing information has been described as a major cause of medication events (14).
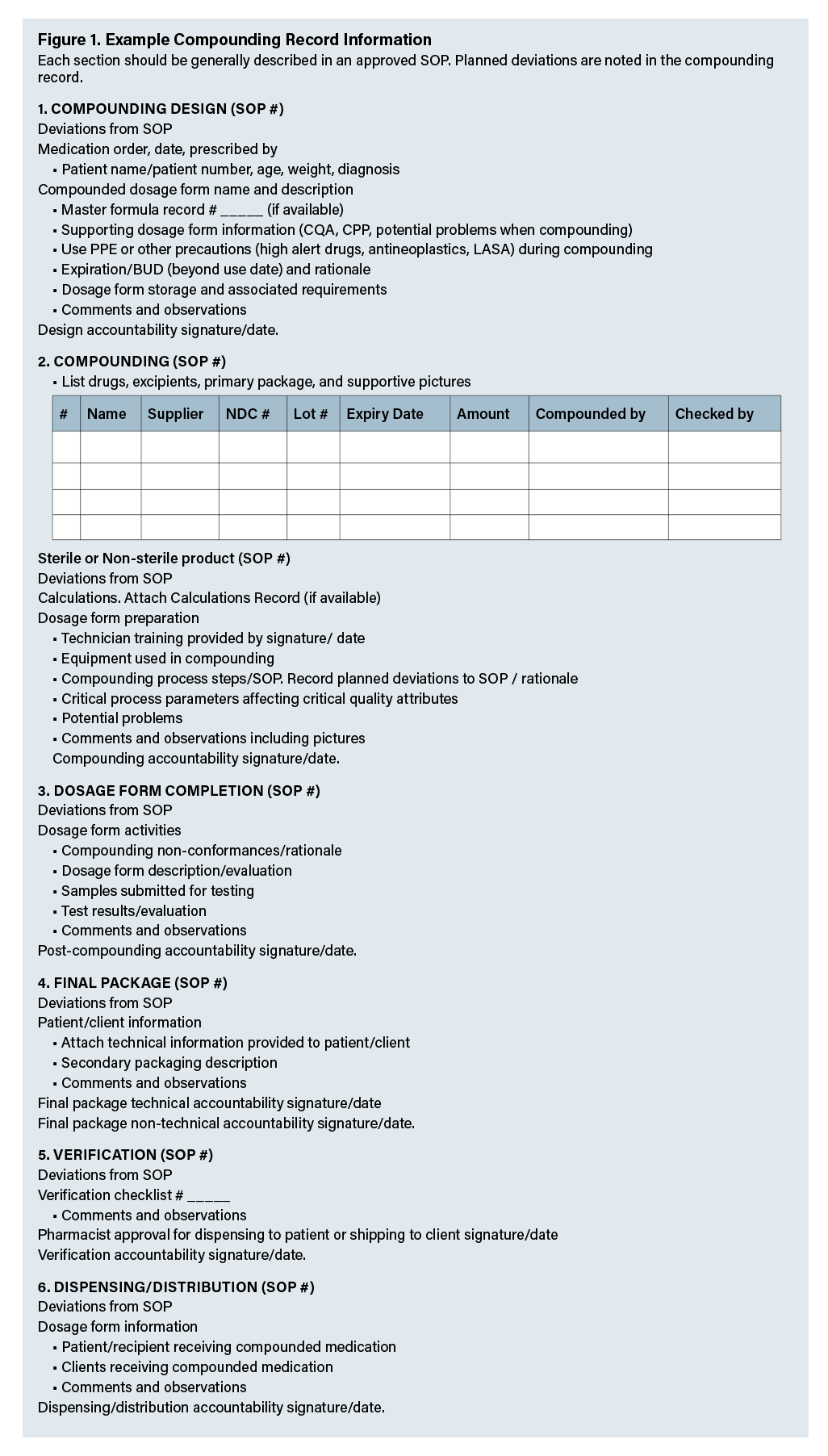
- Patient information. Patient information, such as patient age, weight, medical diagnosis, hepatic function, kidney function, concurrent drug therapy, and other patient factors affecting formulation and dosage, must be reviewed. Is the medication order consistent with diagnosis (e.g., methotrexate dosage for cancer vs. rheumatoid arthritis)?
- Drug, excipients, primary package, and dosage. Textbooks with example formulations provide the basis for pharmaceutical compounding (15-17). Drugs and excipients to be used in compounding must be reviewed. FDA published a list of bulk drug substances that can be compounded (18,19); these must comply with USP/NF standards (20-24). Compounding must not include drugs withdrawn for reasons of safety or efficacy (25). Relevant physicochemical properties, solubility, stability, compatibility with excipients, and other factors related to formulation design are reviewed. Commercial products must be clearly identified (e.g., which heparin dosage strength will be used for dilutions (26)). Inactive formulation ingredients may be problematic in certain patients (e.g., lactose intolerance, dye allergy). The primary package for the compounded dosage form may be a polymeric IV bag, plastic syringe, glass bottle, or other container, some of which may be incompatible or require extra precautions (e.g., IV nitroglycerin requires a non-PVC administration set). Difficult-to-compound dosage forms identified by FDA should not be compounded (27). Biologic drugs are not compounded (28).
- Other technical. Information relevant to labeling, patient instructions, secondary packaging, supply chain shipping (e.g., refrigeration, non-freezing, light protection), and related considerations is assembled. Dosage form labeling is prepared. Medication information for distribution to the patient or to 503B clients is compiled. Assembled information must consider the entire compounding process, including patient use.
Dosage form design and calculations. The compounded dosage form to be prepared is defined based on the medication order and assembled information. Formulation ingredients, process steps, primary package, and relevant information that includes details must be clearly stated in compounding documentation. Exact formulation amounts and process steps that include potential causes of failure are critical. Each medication order should be viewed as a new order and not copied from previous documents; prior calculations must not be assumed to be correct. A specific calculations process has been described (29); calculations may also be incorporated within comprehensive compounding documentation. Calculations must be carefully executed and include written units of measure; omitting descriptive units when calculating is a common cause of error. Problematic notations (e.g., mcg, µg, µ) must be clarified. Calculations should be reviewed by a second person; verification must not be cursory (i.e., the originator is a boss or someone who “never” makes mistakes). Calculations logic, execution, and supporting information must be critically reviewed and include being watchful for number transpositions.
Documentation. The compounding record, work order, batch record, etc. is the permanent record of the compounded preparation retained at the compounding site. Each compounded preparation requires some form of compounding record; a compounded IV solution with a single IV additive prepared according to an SOP will require much less detail than an oral capsule formulation with multiple ingredients and a multi-step compounding procedure. Hospital IV solutions may be recorded within a pharmacy management system or other electronic record (e.g., Epic, DoseEdge). A site SOP should provide documentation templates; a standardized template will increase process uniformity and prevent omissions and errors. The compounding record will be utilized throughout all stages of the compounding process. SOP references are noted for standardized activities; planned deviations are noted in the compounding record. Documentation may contain descriptive information, rationale, ingredients, sources, lot numbers, equipment, process information, references, labeling, patient counseling, and other content; its level of detail must be sufficient to enable an exact repeat preparation of the compounded dosage form. Document clarity is essential; multiple personnel will utilize the compounding record throughout the process. Documentation must not contain drug name abbreviations, problematic units of measure, and numeric problems, such as leading and trailing zeros; awareness of look-alike sound-alike (LASA) drug names (e.g., tetracycline HCl and tetracaine HCl) and other sources of confusion should be prospectively highlighted. Evaluation criteria used post-compounding are identified. A listing of representative compounding record topics is presented in Figure 1. Signature/date of individuals associated with respective content throughout the compounding process is recorded in the compounding record. Compounding records are used for material traceability, adverse event investigations, skills training, and other applications. Some organizations develop a library of master formulation records for frequently prepared dosage forms.
Supporting activities design
- Stage 2: pre-compounding. Stage 2 activities are also designed in stage 1. These include compounding planning followed by preparation of the compounded dosage form. Precautions for problematic activities are highlighted. Critical process techniques must be reviewed with technicians prior to compounding (e.g., particle size reduction, geometric mixing, levigation, syringe use, etc.); additional training may be necessary. Certain drugs and excipients (e.g., potent drugs, antineoplastics, hormones, peanut oil, allergens, beta-lactams (penicillins)) must not contaminate subsequent compounding and require dedicated or disposable equipment. Personal protective equipment (PPE) may be required for technician protection. Potent and dangerous drugs may require restricted access barrier systems or controlled pressure facilities. Stage 2 activities should be generally described in approved site SOPs; planned deviations are noted in the compounding record.
- Stage 3: dosage form completion. Stage 3 activities are also designed in stage 1. These include physical evaluation of the dosage form (e.g., appearance, solution clarity, suspension dispersibility, topical cream absence of particulates, color, and other visuals). Dosage form evaluation must be rigorous and consistent with design expectations. Testing is specified in the compounding record; 503B preparations are submitted for analytical laboratory testing, sterility testing, endotoxin, and other GMP requirements; validated test methods are required. The label for the final dosage form is prepared; labeling must be accurate (e.g., codeine vs. codeine phosphate) without spelling or typo errors, especially in the patient’s name. The compounded dosage form is quarantined to prevent release pending dosage form approval in stage 5. Equipment and facilities must be properly cleaned per SOP; cleaning must consider physical properties of ingredients (i.e., hydrophilic vs. hydrophobic). Dedicated equipment must be identified and stored to prevent inadvertent use; use of disposables eliminates equipment cleaning issues. Drugs or ingredients requiring specialized waste disposal are identified. These activities should be described in approved SOPs; additional requirements are added to the compounding record.
- Stage 4: final package. Stage 4 activities include final requirements for dosage form release to patients, clients, or other recipients. Technical information for patients and clients comprises labeling, dosage, administration, verbal counseling, storage conditions, and other relevant communication. Patient information should be equivalent to that provided by pharma manufacturers for commercial products. The final package provided to the patient/client must ensure correct use of the compounded dosage form. Most activities should be identified in SOPs; deviations are noted in the compounding record. Specialized shipping or storage requirements beyond SOP stipulations are noted.
Stage 1 hazards. Table II provides examples of potential stage 1 hazards, risks, and failure modes (i.e., what might go wrong). Most critical among these are identifying critical dosage form characteristics, assembling and verifying information critical to compounding, calculation errors caused by omitting units of measure in equations, and compounding directions that are too general and non-specific. The point of Table II is to encourage prospective thinking about potential problems; personnel designing the compounded dosage form should anticipate problems and include preventive actions in compounding instructions.
Stage 1 accountability. Signature/date of stage 1 responsible personnel is noted in the compounding record. Personal signature/date affirms responsibility for performance of stage 1 activities.
Stage 2 compounding
Stage 2 compounding comprises pre-work and actual preparation of the compounded dosage form per stage 1 directions. Personnel involved in actual compounding must also plan their activities for expeditious performance, especially when compounding sterile dosage forms.
Pre-compounding. Pre-compounding activities include selecting and organizing drugs, materials, equipment, and supplies for efficient dosage form preparation. Personnel who prepare the compounded formulation must have good understanding of compounding techniques before initiating compounding. Activities involved may be conducted in a non-sterile compounding area, laminar air-flow workstation (LAFW) in a clean room environment, or other suitable controlled conditions. Compounding personnel must be dressed appropriately, including scrubs, sterile gowns, gloves, face masks, and personal protective equipment as required.
Training. Technician training on preparatory skills and safety considerations is conducted as needed. Technical skills to prepare compounded dosage forms will vary depending on type and complexity. Sterile dosage form compounding is high risk due to potential contamination. Non-sterile compounding requires familiarity with preparatory techniques for specific dosage forms.
- Facilities. Preparations containing potent or toxic injectable drugs such as anti-neoplastics may require the use of dedicated clean rooms with differential air pressure monitoring.
- Equipment and dedicated equipment. Compounding equipment must be carefully examined to ensure cleanliness before use. Certain drugs and excipients may require use of dedicated equipment (e.g., potent drugs, hormones, peanut oil, allergenic materials, beta-lactam (penicillin) drugs, and other ingredients that must not contaminate compounded dosage forms).
- PPE. PPE beyond usual garb/equipment may be required for technician safety when working with high-alert, oncology, or other hazardous drugs.
- Material selection and evaluation. Drugs, excipients, and required materials to be used in compounding are assembled. Compounding personnel must be watchful for similar drug names (LASA), standardized packaging, small fonts on labels, and other features potentially contributing to erroneous material selection. Assembled drugs, excipients, and required materials must be critically examined for evidence of stability issues, contamination (e.g., black specks), discoloration, insoluble material in solutions, package integrity, mold growth, expired dating, and other problems (Figure 1). Pictures of final selected materials for inclusion in permanent documentation are recommended.
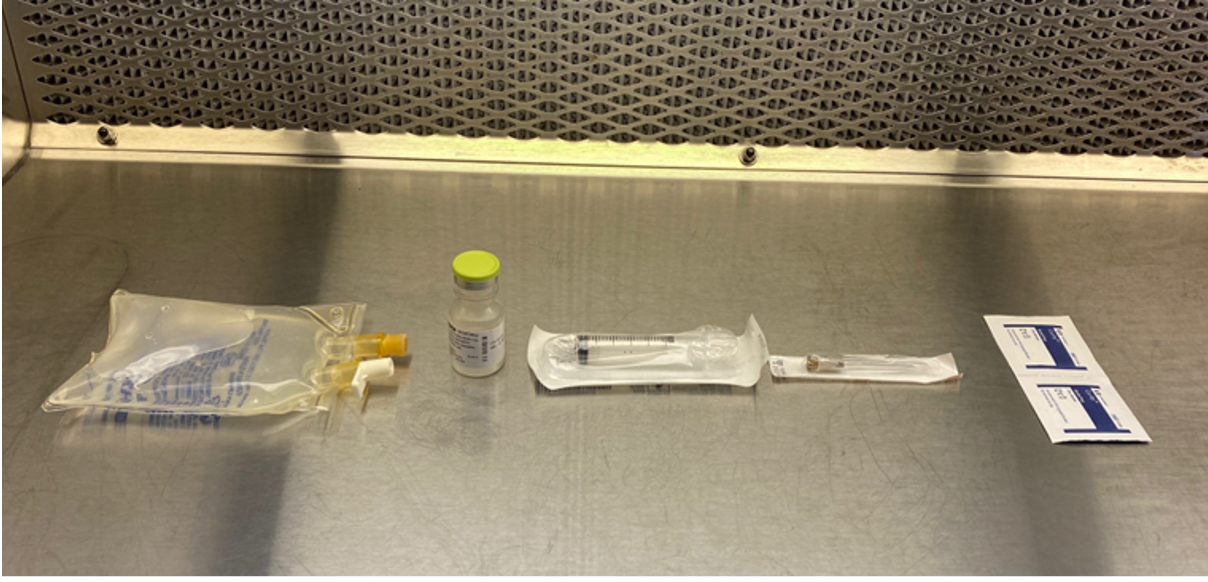
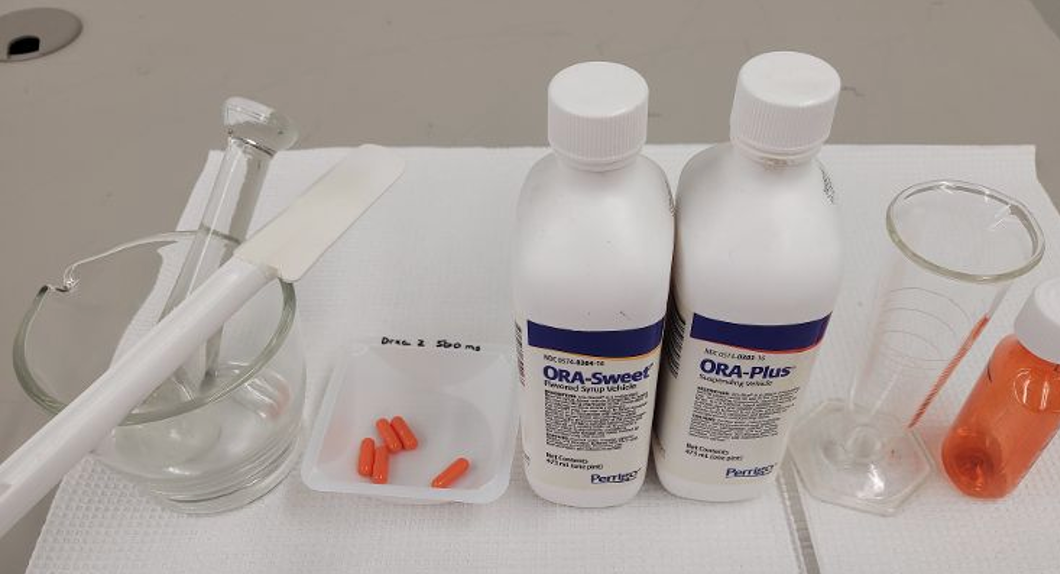
- Set-up. Compounding sterile formulations requires aseptic gowning, sterile needles and syringes, alcohol swabs, and other supplies. Figure 2 illustrates a typical arrangement of sterile compounded formulation components in an LAFW. Properly executing planned activities will ensure expeditious preparation of sterile preparations, minimizing potential contamination. Real-time verification of problematic process steps is critical to ensure correct preparation of sterile dosage forms. Contemporaneous observation of sterile compounding through imbedded workstation cameras is recommended; digital photographs and other technology may also be utilized. The Institute for Safe Medication Practices (ISMP) has long discouraged the “syringe pull back method” to verify sterile product compounding (31). Compounding non-sterile formulations includes selection of ingredients, equipment, and other supplies. Equipment must be qualified, clean, and otherwise suitable for use. Figure 3 illustrates a typical arrangement of non-sterile compounded formulation components. Real-time observation of problematic process steps is especially critical to ensure correct preparation of non-sterile compounded dosage forms; video, digital photographs, or other technology are useful for permanent records.
Dosage form preparation. The designed dosage form is then prepared. The formula, compounding procedure, and documentation as designed in stage 1 are executed. Sterile preparations require attention to aseptic techniques; non-sterile preparations must utilize technical processes specific to the individual dosage form.
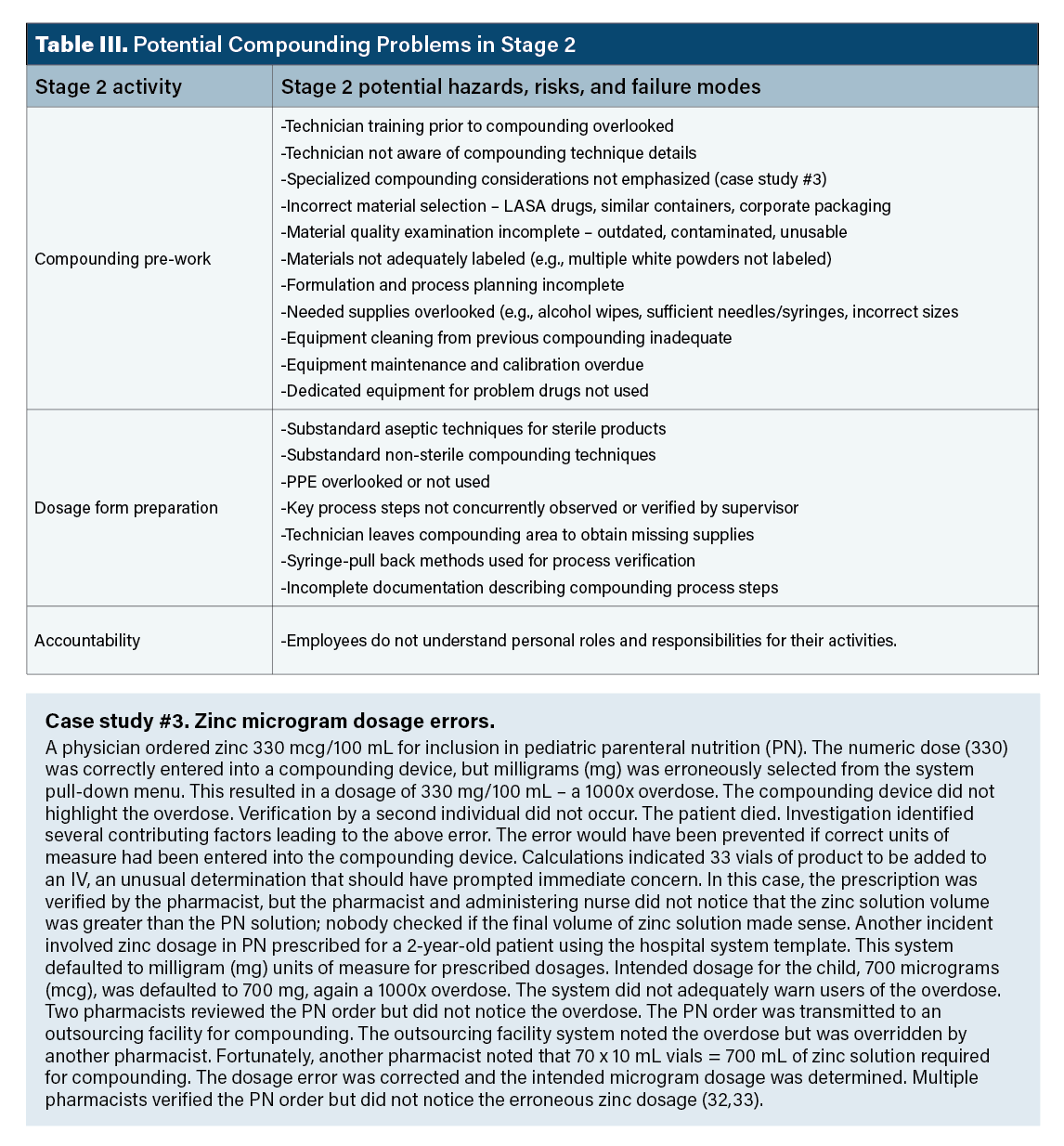
Stage 2 hazards. Table III provides examples of potential hazards occurring in stage 2. The point of Table III is prospective thinking about potential problems. Most critical among these are insufficient planning of compounding activities, overlooking materials used during compounding necessitating retrieval during compounding, substandard compounding skills (especially with less-frequent or unfamiliar techniques), and aseptic technique “shortcuts.”
Stage 2 accountability. Signature/date of stage 2 responsible personnel is noted in the compounding record. Signature/date affirms personal responsibility for performance of stage 2 activities.
Figure 2. Intralipid 20% with damaged packaging (note black oxygen sensor), undissolved mannitol crystals, and contaminated lactose (black specks).
Figure 3. Sterile Product Compounding Planning
Figure 4. Non-Sterile Product Compounding Planning
Stage 3 dosage form completion
Stage 3 post-compounding comprises testing, other dosage form-related activities, and ancillary activities executed after completion of compounding.
Post compounding. Activities directly related to the compounded dosage form described in site SOPs are completed. Deviations to SOPs are identified in the compounding record. Most important among these are the critical evaluation of the compounded preparation, submission of samples for testing, and completion of compounding documentation.
- Sampling and testing. The compounded preparation is evaluated. Visual examination of appearance, color and clarity of solutions, suspension dispersability, topical cream smoothness, absence of solids, freedom from visible foreign particulate matter, package integrity, signs of instability, and other quality attributes are evaluated; physical observation evaluation must be rigorous. 503B compounding samples are submitted for analytical testing to verify potency, sterility, stability, and other requirements identified in stage 1; testing must use validated test methods.
- Technical review. Deviations, non-conformances, environmental monitoring, and other data related to the compounded dosage form are reviewed. Acceptable results are required for eventual dispensing to patients and shipping to clients.
- Documentation. Compounding record documentation is completed. Compounding documentation must ensure full traceability of ingredients, processes, and equipment, and it must provide sufficient detail to enable an identical repeat preparation of the compounded dosage form. Documentation for sterile preparations is completed outside the LAFW.
- Labeling. The compounded dosage form is labeled and associated technical information assembled per stage 1 directions.
- Quarantine. The compounded dosage form is quarantined until all testing is completed and reviewed.
Ancillary activities. Ancillary activities include workspace and equipment cleaning and waste disposal. If dedicated equipment is used for specific ingredients, it must be cleaned, dried, labeled, and appropriately stored to prevent future inadvertent use; misuse of dedicated equipment is a common compounding error. Waste disposal must follow site procedures (i.e., “sharps,” recyclables, HIPAA, biologicals, oncology drugs, and drug waste disposed into appropriate containers). Routine post-compounding activities are completed per SOPs. Deviations from SOPs are noted in the compounding record.
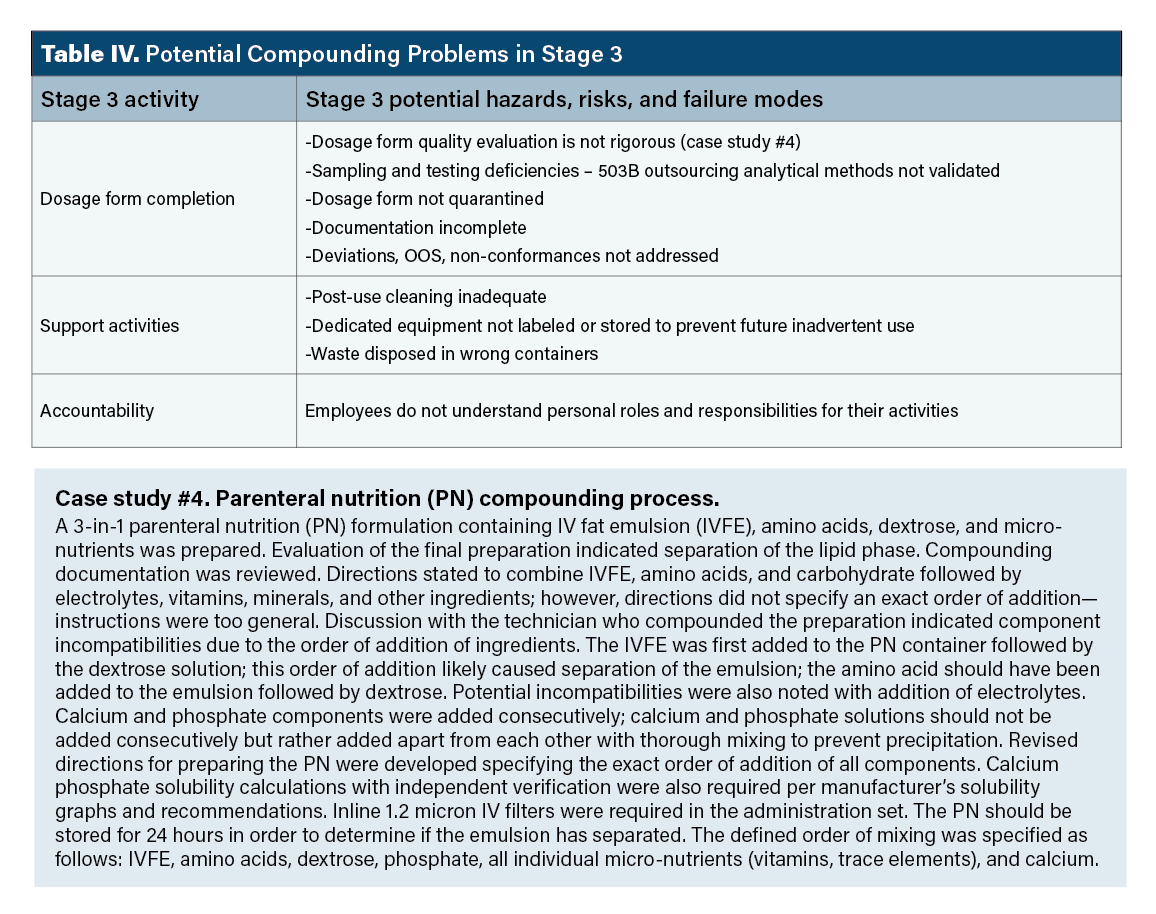
Stage 3 hazards. Examples of possible stage 3 hazards are presented in Table IV. The point of Table IV is prospective thinking about potential problems. A common stage 3 problem is dosage form evaluation; preparations must strive for excellence and not be just “good enough.” Equipment used in compounding must be scrupulously cleaned after use. Dosage forms must be quarantined to prevent release until final approval (stage 5),
Stage 3 accountability. Signature/date of stage 3 responsible personnel is recorded in the compounding record. Signature/date affirms personal responsibility for stage 3 activities.
Stage 4 final package
Stage 4 final package comprises technical and associated activities that complete dosage form preparation. Supportive information for patients or clients is compiled as specified in stage 1.
Technical. Test results are compiled and evaluated. Failing or unexpected test results must be investigated, appropriate action taken, and corrective action implemented. Labeling, medication information, patient counseling, and other communication to be provided to patents and clients is compiled. Commercial product manufacturers provide medication information to patients; comparable information should be developed on patient/client interactions. ISMP Medication Learning Guides for Consumers exemplify drug information provided to patients at dispensing (34). The compounded dosage form must be used correctly by the patient to ensure efficacy.
Associated activities are identified in an SOP or specified in the compounding record. Deviations to SOPs may include specialized protective packaging, time/temperature limitations with tracking devices, and other shipping requirements. Shipping should be performed by a dedicated delivery service in a controlled manner with location-tracking capability; shipping must include temperature control.
Documentation. Compounding record documentation is finalized. This is the final compounding record retained by the organization. After the compounded dosage form is dispensed or shipped, the compounding record is the lasting evidence of compounded dosage form preparation.
Stage 4 hazards. Examples of possible stage 4 hazards are presented in Table V. The point of Table V is prospective thinking about potential problems. Most important among these are assembly of communication for patients and clients. The compounded dosage form must be used properly by the patient, administered correctly, and otherwise handled to be efficacious. Completion of documentation is also critical in stage 4. Compounded dosage form quarantine (stage 3) should prevent inadvertent release to patients or clients.
Stage 4 accountability. Signature/date of stage 4 responsible personnel is recorded in the compounding record. Signature/date affirms personal responsibility for stage 4 activities.
Stage 5 verification
Stage 5 verification is the final check on the compounded dosage form, packaging, documentation, and related information before dispensing to the patient or shipping to clients.
Review and evaluation. Review of the completed dosage form and associated elements is a critical step in the compounding process. Ideally, verification is accomplished by an independent observer separate from people involved in the compounding process. Verification addresses the final dosage form, test results, completed documentation, and associated communications to be provided to patients or clients. The compounded dosage form is not dispensed to patients or shipped to clients until the final compounded dosage form and associated information are approved.
Review by a competent, independent observer must be a serious and thorough activity by an impartial observer—not a perfunctory ”rubber stamp” exercise. ISMP has discussed the judicious use of independent double-checks in pharmacy practice (35). Final dosage form and associated information must be reviewed vs. the original medication order. Review must be completed before the compounded dosage form is dispensed or shipped. Checklists are useful to ensure a comprehensive review. Errors, omissions, or insufficient details in compounding documentation are corrected. Documentation will be utilized in future investigations, training, and other applications. Final documentation is retained in prescription files, patient records, manufacturing batch records, or electronic systems. An example checklist with relevant topics is provided in Figure 5.
Compounded dosage form approval. When dosage form appearance, test results, documentation, and other reviewed content are acceptable, the compounded dosage form is approved for release by the responsible pharmacist. Approval is documented in the compounding record. The dosage form is removed from quarantine.
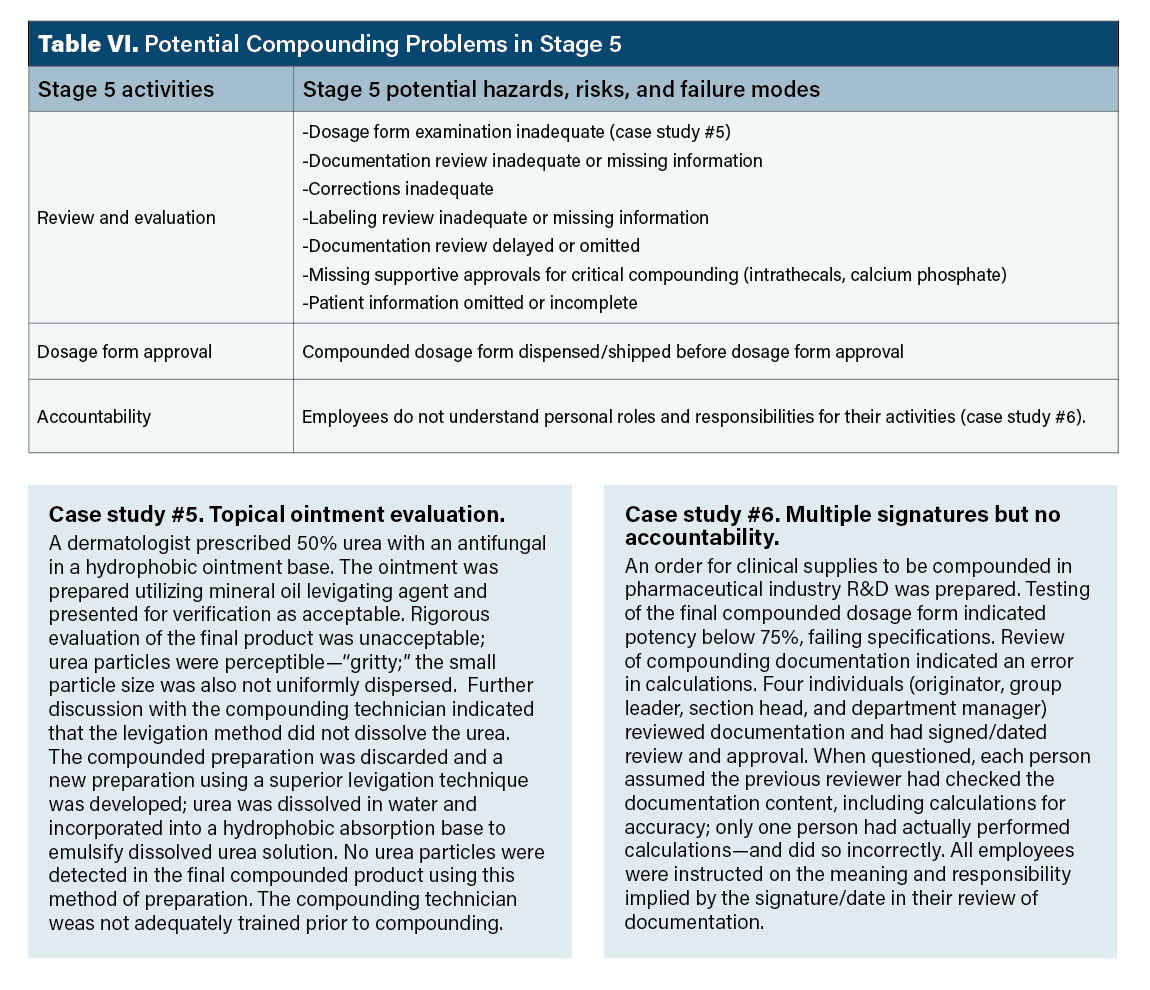
Stage 5 hazards. Examples of possible stage 5 verification hazards are presented in Table VI. The point of Table VI is prospective thinking about potential problems. Potential hazards in stage 5 are critical; this is the final check of dosage form quality, test results, patient/client documentation, and compounding record documentation. Compounding record documentation is the lasting record of the compounding process.
Stage 5 accountability. Signature/date of stage 5 responsible personnel is recorded in the compounding record. Signature/date affirms personal responsibility for stage 5 activities.
Stage 6 dispensing/distribution
Stage 6 comprises activities in which compounded dosage forms are provided to users.
Dispensing/shipping to patients/clients. Stage 6 includes dosage form dispensing to patients, providing drug information, and counseling as needed. Dispensing and counseling are performed according to the clinical situation and patient needs. Stage 6 also includes shipping to patients per secondary package design, including temperature control, light protection, package tracking, and other requirements. Approval to dispense/ship is documented in stage 5; dispensing or shipping must not occur before the responsible pharmacist has provided final dosage form approval. Stage 6 also includes shipping to clients per secondary package design after responsible pharmacist approval.
Numerous examples exist of drug products being dispensed to the wrong patient and conditions under which this may occur. Errors may occur in community settings or in the hospital environment for a variety of reasons. Obvious causes involve patients with identical last names. Hospitalized patients may switch beds without notifying the nursing staff. Electronic order entry may be confused; mis-transcribed telephone orders, dispensing system overrides, and other potential patient mixups may occur. Verification using two patient identifiers is recommended to confirm the intended patient as well as verify correct treatment to the individual. These actions should occur at each step in the medication process (i.e., from original medication order, through multiple transcriptions {physicians, pharmacists, nurses}, and final dosage form administration to the patient). Examples of questions to be asked before drug administration, dispensing, or counseling to verify they are the intended patient typically include the patient’s name, date of birth, last four digits of the US social security number, or other government identification. Only verifying the patient’s name is not sufficient. Patients for whom English is a second language require increased assurance that they are the intended patient.
Shipping to clients. Stage 6 also includes shipping to clients per secondary package design. Approval to dispense/ship is documented in Stage 5; again, dispensing or shipping must not occur before the responsible pharmacist has provided final dosage form approval.
Stage 6 hazards. Examples of possible stage 6 hazards are presented in Table VII. The point of Table VII is prospective thinking about potential problems. Most significant among these are dispensing a drug to the wrong patient. This may occur in community and hospital settings; there are numerous opportunities for actions that contribute to dispensing/administration of dosage form to the wrong patient (36-38).
Stage 6 accountability. Signature/date of stage 6 responsible personnel is noted in the compounding record.Signature/date affirms personal accountability for stage 6 activities.
Management responsibilities
Management of a compounding organization is responsible for daily compounding performance, as well as related supportive activities. Quality systems guidance documents identify expectations of management (e.g., leadership, structure, quality system design, policies, objectives, plans, system review). FDA issued CDER’s Quality Management Maturity Program (39) addressing expectations beyond current GMP; compounding management must aspire to much more than minimal GMP compliance. Development of a quality culture mindset in which risks to compounded product quality are minimized is led by management (40)and should be forefront in the organization. ICH Q9(R1) QRM addresses “What might go wrong?” and describes common methods of risk assessment; these methods are directly applicable to pharmaceutical compounding.
Primary management responsibilities in compounding organizations include the following:
- Personnel. Hiring capable personnel is the most important function of management. Compounding personnel must have appropriate technical compounding skills; organizational skills, self-discipline, attention to detail, language skills, legible handwriting, common sense, and realization of personal deficiencies (i.e., knowing what you don’t know) are desirable. Interpersonal skills in a teamwork environment are essential. Sufficient number of personnel must also be hired commensurate with workload.Management is also responsible to assure that each staff member knows and understands their roles and responsibilities, as well as accepts personal responsibility for performance.
- Plans, policies, and procedures. Management is responsible for the standards that define daily workplace activities. Compounding organizations must have written and approved documents; compliance with these documents is mandatory. Inadequate procedures, training, and non-compliant performance are always among the most frequent GMP violations in pharma (41). SOP compliance must be forefront in compounding organization performance. SOPs should be living documents, periodically reviewed for accuracy and relevance and updated when new information is available.
- Training. Personnel must be trained and periodically re-trained on SOPs. Training must be documented; if not documented, it didn’t happen. Training must emphasize personal accountability for SOP requirements; personnel must understand that their signature/date in compounding documentation affirms personal responsibility.
- Communication. The workplace environment must encourage communication of problems, mistakes, and issues without fear of punishment or embarrassment. Management must develop a collaborative relationship with staff with ongoing focus on quality (i.e., a true quality culture that is evident in personnel performance). ISMP identified communications about medication safety risks occurring within and outside the organization as best practices for community (42) and hospital pharmacy (43). FDA-483 observations and warning letters demonstrating compliance deficiencies in compounding organizations are readily available (44).
- Commitment. Beyond hiring, procedures, and training are the everyday expressions of management commitment to quality. Management focus must expand from traditional management to include anticipation of performance hazards and risks (i.e., what might go wrong). Management daily performance in routine activities must exemplify commitment to quality.
- Administrative. Management is responsible for administrative functions supportive to compounding (e.g., funding, scheduling, maintenance, calibration, facility cleanliness).
Management hazards.Examples of possible management hazards are presented in Table VIII. Most significant among these are deficiencies in management commitment to responsibilities; organization culture is directly related to management commitment. The actions of individual employees reflect the attitude and commitment of management—what management wants, management gets. Management is also ultimately responsible to address potential hazards identified above in respective stages of the compounding process.
Management accountability. Signature/date of management on policies, procedures, and other documents affirms management accountability.
Summary
This discussion describes an ordered process for pharmaceutical compounding based on academic teaching of compounding theory and practice, problem experiences in multiple settings, regulatory observations, and published medication errors. Technical activities occurring in the compounding workplace beyond physical compounding techniques are addressed. The process describes what and why activities, as well as what might go wrong throughout the comprehensive process.
Six stages of compounding activities are identified, each having specific objectives. Stage 1 design activities address considerations to be executed in all subsequent compounding stages; technical consistency throughout the process is forefront. Compounding activities are executed in stages 2, 3, and 4. Stage 2 comprises selection of materials and equipment, compounding planning, and actual dosage form preparation. Stage 3 post-compounding comprises dosage form review, sampling, testing, and related activities. Stage 4 includes final review of test results; medical information and packaging for dispensing to patients or shipping to clients is compiled. Stage 5 verification comprises review of the compounded dosage form, labeling, drug information, and secondary packaging; stage 5 activities require an independent observer. The compounded medication is approved for dispensing in stage 5. Stage 6 dispensing/distribution provides the compounded dosage forms to users, including patient counseling, medical information, and/or appropriate supply chain shipping. Compliance with regulations and site procedures is required throughout the process. Personnel understanding of their responsibilities and affirming accountability by signature/date on documentation is required. Examples of potential hazards are identified throughout; case studies further demonstrate problematic circumstances.
Management has significant technical responsibilities in the compounding process. Management is responsible for the actual compounding, as well as for supportive functions. Management must continually support employee training and awareness of possible problems. As risks are identified and knowledge is gained, SOPs are modified, including corrective and preventive actions for continuous improvement. A workplace environment in which quality is forefront (i.e., a quality culture) must be an ongoing goal of compounding management and staff.
References
- Pluta, P.L. Pharmaceutical Compounding Overview – Technical, Regulatory, and Management in a Workplace Environment. Pharmaceutical Technology 2024, 48(2) 18-22. Compounding Overview: Primary Considerations for the Workplace (pharmtech.com). (accessed 7-1-25)
- FDA. Drug Shortages, https://www.accessdata.fda.gov/scripts/drugshortages/default.cfm (accessed 7-20-24).
- FDA. Compounding Certain Ibuprofen Oral Suspension Products Under Section 503B of the Federal Food, Drug, and Cosmetic Act. February, 2023. https://www.fda.gov/media/164693/download (accessed 7-1-25).
- Yu, L. X. Pharmaceutical Product Quality, Quality by Design, cGMP, and Quality Metrics. FDA. 01-yu-fda-pqri-2015-ver1.pdf (accessed 7-1-25).
- Yu, L.X., G. Amidon, M.A. Khan, S.W. Hoag, J. Polli, G.K. Raju, and J. Woodcock. Understanding Pharmaceutical Quality by Design. MIT Open Access Articles. AAPS J 16, 34, 5-23-2014. 771-783. https://dspace.mit.edu/bitstream/handle/1721.1/106646/12248_2014_Article_9598.pdf?sequence=1 (accessed 7-1-25).
- ICH Q8(R2). Pharmaceutical Development. August 2009. Q8_R2_ (ich.org) (accessed 7-1-25).
- ICH Q9(R1). Quality Risk Management, Q9(R1) 1-18-2023. https://database.ich.org/sites/default/files/ICH_Q9%28R1%29_Guideline_Step4_2023_0126_0.pdf (accessed 7-1-25).
- FDA. Guidance for Industry.Quality Systems Approach to Pharmaceutical CGMP Regulations, September, 2006. https://www.fda.gov/media/71023/download (accessed 7-1-25).
- ICH. Pharmaceutical Quality System Q10. 6-4-2008. https://database.ich.org/sites/default/files/Q10%20Guideline.pdf (accessed 7-1-25).
- American Society of Health System Pharmacists. ASHP Guidelines on Compounding Sterile Preparations https://www.ashp.org/-/media/assets/policy-guidelines/docs/guidelines/compounding-sterile-preparations.ashx (accessed 7-1-25).
- National Association of Pharmacy Regulatory Authorities. Guidance Document for Pharmacy Compounding of Non-Sterile Preparations, 2018. NAPRA-Mdl-Stnds-Pharmacy-Compounding-Nonsterile-Preparations-Guidance-EN-June-2018-CLAR-Jan-2022.pdf (accessed 7-1-25).
- ISMP. ISMP Guidelines for Safe Preparation of Compounded Sterile Preparations, 2016. https://www.ismp.org/sites/default/files/attachments/2017-11/Guidelines%20for%20Safe%20Preparation%20of%20Compounded%20Sterile%20Preperations_%20revised%202016.pdf (accessed 7-1-25).
- Campbell, Robert. The Joint Commission’s Medication Compounding Certification Program, 2017. https://www.pppmag.com/article/2068#:~:text=Campbell%3A%20The%20Joint%20Commission%20Medication%20Compounding%20Certification%20is,%3C797%3E.%20Certification%20is%20not%20offered%20for%20outsourcing% (accessed 7-20-24).
- Shaw, Gina. Missing Information a Top Cause of Medication Events. Pharmacy Practice News, 7-26-24. https://www.pharmacypracticenews.com/Operations-and-Management/Medication-Safety/Article/07-24/Missing-Information-a-Top-Cause-of-Medication-Events/74203 (accessed 7-1-25).
- Allen, Lloyd V., Jr. The Art, Science, and Technology of Pharmaceutical Compounding, 6th ed. American Pharmacists Association, Washington, DC, 2020.
- Allen, Lloyd V, Jr. International Journal of Pharmaceutical Compounding. https://ijpc.com/Index.cfm (accessed 7-1-25).
- Shrewsbury, Robert. P. Applied Pharmaceutics in Contemporary Compounding, 3rd ed. Morton Publishing, Englewood, CO 80110) 2015.
- FDA. 21 CFR §216.23. Bulk drug substances that can be used to compound drug products in accordance with section 503A of the Federal Food Drug, and Cosmetic Act. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-216/subpart-B/section-216.23 (accessed 7-1-25).
- FDA. 503B Bulks List. 8-21-2023. (accessed 7-20-24)
- USP Pharmaceutical Compounding — Non-Sterile Preparations, 1-11-22. https://www.usp.org/compounding/general-chapter-795 (accessed 7-1-25).
- USP Pharmaceutical Compounding — Sterile Preparations, 1-11-22. https://www.usp.org/compounding/general-chapter-797 (accessed 7-1-25).
- USP Hazardous Drugs – Handling in Healthcare Settings. https://www.usp.org/compounding/general-chapter-800 (accessed 7-1-25).
- USP Quality assurance in Pharmaceutical Compounding. https://www.drugfuture.com/Pharmacopoeia/USP32/pub/data/v32270/usp32nf27s0_c1163.html#google_vignette (accessed 7-1-25).
- USP Compounding Compendium. https://www.usp.org/products/usp-compounding-compendium (accessed 7-1-25).
- FDA. 21 CFR §216.24. Drug products withdrawn or removed from the market for reasons of safety or effectiveness. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-216/subpart-B/section-216.24 (accessed 7-1-25).
- Arimura, J., R.L. Poole, M. Jeno, W. Rhine, and P. Sharek. Neonatal Heparin Overdose—A Multidisciplinary Team Approach to Medication Error Prevention. J.Pediatr Pharmacol Ther. 2008 Apr-Jun 13(2); 96-98. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3462065/ (accessed 7-1-25).
- FDA. Drug Products or Categories of Drug Products that Present Demonstrable Difficulties for Compounding under Sections 503A or 503v of the federal Food, Drug, and Cosmetic Act, 3-20-24. https://www.federalregister.gov/documents/2024/03/20/2024-05801/drug-products-or-categories-of-drug-products-that-present-demonstrable-difficulties-for-compounding (accessed 7-1-25).
- FDA. Compounding and the FDA: Questions and Answers. 6-29-22. https://www.fda.gov/drugs/human-drug-compounding/compounding-and-fda-questions-and-answers#:~:text=Can%20biologics%20be%20compounded%3F%20No.%20Biological%20products%20are,the%20scope%20of%20an%20approved%20biologics%20license%20application (accessed 7-1-25).
- Pluta, P.L., A. Mancini, N.B. Thakar, and V. Chaiyaperm. Dosage Form Calculations Process in a Workplace Environment. Pharmaceutical Technology. Trends in Manufacturing ebook, 5-2024, 22-34. https://www.pharmtech.com/view/pharmaceutical-compounding-calculations-in-a-workplace-environment (accessed 7-1-25).
- ISMP. Life-threatening Errors with Flecainide Suspension in Children. 4-23-2015. https://www.ismp.org/sites/default/files/attachments/2018-03/community201504.pdf (accessed 7-1-25).
- ISMP. Maximize Benefits of IV Workflow Management Systems by Addressing Workarounds and Errors. 9-7-17. https://www.ismp.org/resources/maximize-benefits-iv-workflow-management-systems-addressing-workarounds-and-errors (accessed 7-1-25).
- ISMP. Too Close for Comport; Fatal zinc overdose narrowly avoided. 7-4-19. https://www.nutritioncare.org/uploadedFiles/Documents/Newsletter/ISMP%20Newsletter%20July%204%202019%20-%20near%20miss%20zinc%20overdose.pdf (accessed 8-1-24).
- Grissinger, Matthew. A Fatal Zinc Overdose in a Neonate. P&T, Jul 36(7), 2011. https://ncbi.nlm.nih.gov/pmc/articles/PMC3171817/ (accessed 7-1-2).
- ISMP. High-Alert Medication Learning Guides for Consumers, 11-2-18. https://www.ismp.org/resources/high-alert-medication-learning-guides-consumers (accessed 7-1-25).
- ISMP. Independent Double Checks: Worth the Effort if Used Judiciously and Properly. 6-6-2019. https://www.ismp.org/resources/independent-double-checks-worth-effort-if-used-judiciously-and-properly (accessed 5-1-24).
- Yang, Annie and M. Grissinger. Wrong-Patient Medication Errors: An Analysis of Event Reports in Pennsylvania and Strategies for Prevention. Pennsylvania Patient Safety Advisory, Vol 10, #2, June 2013. https://patientsafety.pa.gov/ADVISORIES/Documents/201306_home.pdf (accessed 7-1-25)
- Schulmeister, Lisa. Patient Misidentification in Oncology Care. Clinical Journal of Oncology Nursing, V 12, #3, June 2008. https://pubmed.ncbi.nlm.nih.gov/18515248/ (accessed 7-1-25).
- Grissinger, Matthew. Oops, Sorry, Wrong Patient. P&T 39(8), 535-537, Aug 2014. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4123800/ (accessed 7-1-25).
- FDA. CDER Quality Management Maturity. 1-26-24. https://www.fda.gov/drugs/pharmaceutical-quality-resources/cder-quality-management-maturity#:~:text=FDA%E2%80%99s%20Center%20for%20Drug%20Evaluation%20and%20Research%20%28CDER%29,go%20beyond%20current%20good%20manufacturing%20practice%20%28CGMP%29%20requirements (accessed 7-1-25).
- Friedman, Rick. Establishing a Culture of Quality, September, 2021. https://www.fda.gov/media/156357/download?attachment (accessed 7-1-25)
- Hoare, Aaron. The Most Common FDA 483 Observations. https://www.ideagen.com/thought-leadership/blog/the-most-common-fda-483-observations, 4-18-2023 (accessed 7-1-25).
- ISMP. First ISMP Targeted Medication Safety Best Practices for Community Pharmacy Released, 4 Apr, 2023. https://www.ismp.org/news/first-ismp-targeted-medication-safety-best-practices-community-pharmacy-released (accessed 7-20-24).
- ISMP. Best Practice 14 in 2018-2019 Targeted Medication Safety Best Practices in Hospitals. https://www.ismp.org/sites/default/files/attachments/2019-01/TMSBP-for-Hospitalsv2.pdf (accessed 7-1-25).
- FDA. Compounding: Inspections, Recalls, and Other Actions, 4-23-24. https://www.fda.gov/drugs/human-drug-compounding/compounding-inspections-recalls-and-other-actions (accessed 7-1-25).
Acknowledgments
Contributions from Jan M. Keresztes, RPh, PharmD, pharmaceutical educator, Jeanne Moldenhauer, pharmaceutical consultant, Excellent Pharmaceutical Consultants, Richard Poska, RPh, managing director, Flexo CMC Consulting, William R. Porter, PhD, principal scientist, Peak Process Performance Partners, and Chloe Gutierrez, student at Roosevelt University College of Science, Health, and Pharmacy, Schaumberg, IL, are greatly appreciated.
Authors
Paul L. Pluta, RPh, PhD, is a pharmaceutical scientist with pharmaceutical industry, academic teaching, journal editorship, community pharmacy, and hospital pharmacy experience.
Alan M. Mancini, RPh, is a pharmaceutical scientist with pharmaceutical industry, academic teaching, community pharmacy, and hospital pharmacy experience.
Nishant B. Thakar, RPh, PharmD, is Assistant Professor Clinical Sciences, Roosevelt University College of Science, Health, and Pharmacy, Schaumburg, IL, USA, with community pharmacy and hospital pharmacy experience.
Varanya Chaiyaperm, RPh, PharmD is Clinical Assistant Professor University of Illinois College of Pharmacy, Chicago, IL, USA, with community pharmacy and hospital pharmacy experience.